![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
This month's installment of "Validation Viewpoint"outlines how forced degradation is used to develop a stability-indicating method (SIM) and examines some of the method development and validation parameters critical to its success. The Stability-Indicating Method A SIM is a quantitative analytical procedure used to detect a decrease in the amount of the active pharmaceutical ingredient (API) present due to degradation. According to FDA guidelines, a SIM is defined as a validated analytical procedure that accurately and precisely measures active ingredients (drug substance or drug product) free from potential interferences like degradation products, process impurities, excipients, or other potential impurities, and the FDA recommends that all assay procedures for stability studies be stability indicating (3). During stability studies, liquid chromatography (LC) is used routinely to separate and quantitate the analytes of interest. There are three components necessary for implementing a SIM: sample generation, method development, and method validation.
For most samples, storage for as long as two weeks at 80 °C, or six weeks at 60 °C, is used. Samples should be stored in appropriate vessels that allow sampling at timed intervals and that protect and preserve the integrity of the sample. Thermostated and humidity-controlled ovens also should be employed. Generally, the goal of these studies is to degrade the API 5–10 %. Any more than this and relevant compounds can be destroyed, or irrelevant degradation products produced (for example, degradation products of the degradation products). Any less, and important products might be missed. Experience and data obtained from studies performed previously on related compounds also should be used when developing new protocols. Step Two: Developing the LC Method: After the sample is generated through the use of a properly designed and executed forced degradation, it can be used to develop the LC method. These days, LC method development often is performed on gradient systems capable of automated column and solvent switching, and temperature control. Systems and software that automate the process, some with decision making built-in, also have been reported (5). Scouting experiments often are run, and then conditions are chosen for further optimization. Resolving power, specificity, and speed are key chromatographic method attributes to keep in mind during method development. But because LC method development often can be a complex process, a full discourse on LC method development is outside the scope of this column. However, excellent resources are available to anyone not already schooled in the art (6). Instead, in this column, we'll focus on a few highlights and new developments pertinent to developing SIMs.
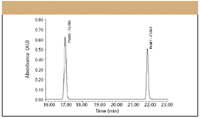
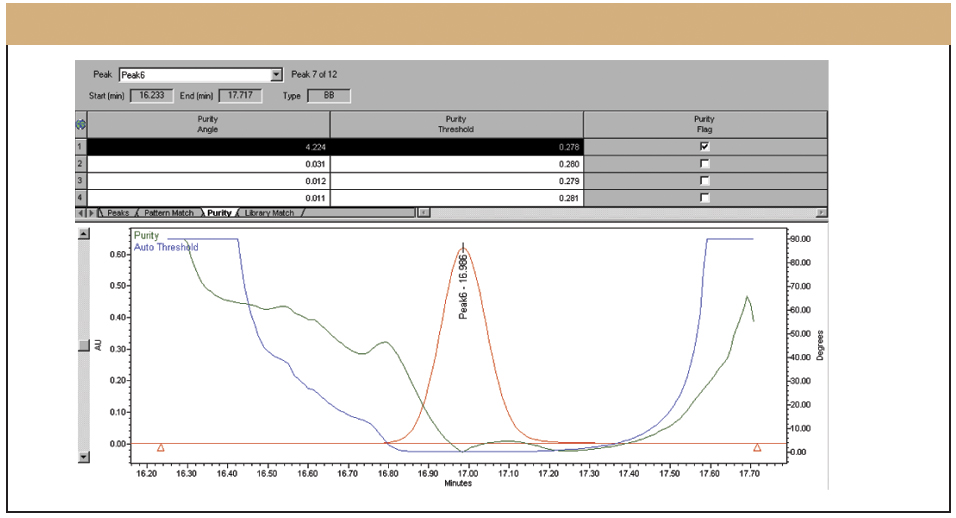
Evaluating Specificity During Method Development Another key parameter to evaluate during method development is specificity. The United Sates Pharmacopeia (USP) and various International Conference on Harmonization (ICH) guidelines define specificity as the ability of a method to assess unequivocally the analyte of interest in the presence of potential interferences (9–11). In the past, it has been acceptable to evaluate resolution, peak shape, and tailing factors to measure and document specificity. However, starting with USP 24, and as a direct result of the ICH process, it was recommended that a peak purity test based upon photodiode-array (PDA) detection or mass spectrometry (MS) be used to demonstrate that a given peak was pure. Modern PDA technology is a powerful tool for evaluating specificity. PDA detectors can collect spectra across a range of wavelengths at each data point collected across a peak, and through software manipulations involving multidimensional vector algebra, they compare each of the spectra to determine peak purity. In this manner, PDA detectors today can distinguish minute spectral and chromatographic differences not readily observed by simple overlay comparisons (12–14). To be successful, three components are required:
An examination of peak 2 indicated the peak was pure. However, a close examination of the spectral information related to peak one reveals a different situation. In Figure 3, the calculated peak purity (in green) is plotted against the noise threshold (in blue), both superimposed on the red chromatographic trace. The purity plot clearly indicates a coelution in the front of the peak as the purity plot exceeds the threshold, as more and more method development work is necessary.
MS has come a long way from the days in which many companies had a dedicated central MS lab and staff. Modern mass spectrometers are smaller, simpler, and operate from the same software used to operate the chromatographic system or other detectors commonly utilized, decreasing the learning curve. MS can provide unequivocal peak purity information, exact mass, and structural and quantitative information depending upon the type of instrument used. MS is also a very useful tool to track peaks as they move around in response to selectivity manipulations in method development. However, only the combination of both PDA and MS on a single instrument and software platform provides the type of valuable orthogonal information required when evaluating specificity and developing SIMs.
A comparable HPLC separation would take 60 min to perform, not including column reequilibration. The ultrahigh pressure LC technique accomplishes the same separation in much less time (4 min) with better resolution and sensitivity. And because it uses the same theories and principles as HPLC, due to the high speed of the analyses, methods can be developed much faster. Step Three: Validate the Method: Different methods have different requirements when it comes to validation. The USP recognizes four method categories and defines the analytical performance characteristics that must be measured to validate each method type (3,9):
Table II summarizes each category and the analytical performance characteristics that must be investigated. SIMs fall into the quantitative division of Category 2, and as such, all analytical performance parameters must be determined except for the limit of detection. (Limit of quantitation would apply instead since SIMs need to be quantitative.) Details of each of the remaining analytical performance parameters (definitions, measurement, and documentation) have been summarized previously in this column and elsewhere and it is not necessary to repeat them here (3,18–21). However, it should be noted that the hyphenated techniques (for example, LC–PDA, LC–MS, and LC–MS-MS) and new technology (for example, ultrahigh pressure LC) outlined previously for method development also can be used to great advantage in method validation. For example, specificity (selectivity), while certainly a goal in method development, also must be demonstrated for proper validation. Therefore, all of the earlier discussionrelating to specificity (selectivity) in method development also would apply to validating the method. Conclusion Proper implementation of a SIM relies on three critical aspects: generation of the sample, and development and validation of the method. The use of a properly designed and executed forced-degradation study will generate a representative sample that will in turn help to ensure that the resulting method adequately reflects long-term stability. New technology and hyphenated advanced detector techniques are extremely valuable in providing increased levels of resolution and specificity and faster analysis times resulting in higher sample throughput. Although specificity does play a central role, all of the remaining pertinent validation parameters also must be evaluated in order to properly validate a SIM in a regulated environment and ultimately ensure the method accomplishes its intended purpose. Acknowledgments The authors would like to acknowledge Michael Jones, Bill Warren, and Uwe Neue of Waters (Milford, Massachusetts) for contributions to this manuscript. References (1) ICH Q1A: Stability Testing of New Drug Substances and Products. (International Conference on Harmonization of Technical Requirements for the Registration of Drugs For Human Use, Geneva, Switzerland, February 2003). See also at www.ICH.org/. (2) ICH Q1B: Stability Testing: Photostability Testing of New Drug Substances and Products (International Conference on Harmonization of Technical Requirements for the Registration of Drugs For Human Use, Geneva, Switzerland, November 1996). See also at www.ICH.org/. (3) Guidance for Industry, Analytical Procedures and Method Validation, U.S. Department of Health and Human Services FDA, August 2000 (www.fda.gov/guidance/index.htm). (4) D.W. Reynolds, Am. Pharm. Rev., May/June, 56–61 (2004). (5) M.E. Swartz, M.D. Jones, and M.A. Andrews, LCGC Application Notebook, 66–67 (June 2002). (6) L. Snyder, J.J. Kirkland, and J. Glajch. Practical HPLC Method Development, 2nd ed. (Wiley-Interscience Publishers, New York, 1997). (7) U.D. Neue, T.H. Walter, B.A. Alden, Z. Jiang, R.P. Fisk, J.T. Cook, K.H. Glose, J.L. Carmody, J.M. Grassi, Y.-F. Cheng, Z. Lu, and R.J. Crowley, Am. Lab. 31, 36 (1999). (8) Y.-F. Cheng, T.H. Walter, Z. Lu, P. Iraneta, C. Gendreau, U.D. Neue, J.M. Grassi, J.L. Carmody, J.E. O'Gara, and R.P. Fisk, LCGC 18(10), 1162 (2000). (9) The United States Pharmacopeia 25/National Formulary 20, Ch. 1225, pg. 2256–2259. (The United States Pharmacopeial Convention, Inc., Rockville, Maryland, 2002). (10) ICH Q2B: Validation of Analytical Procedures: Methodology (International Conference on Harmonization of Technical Requirements for the Registration of Drugs For Human Use, Geneva, Switzerland, May 1997). (11) ICH Q2B: Validation of Analytical Procedures: Methodology (International Conference on Harmonization of Technical Requirements for the Registration of Drugs For Human Use, Geneva, Switzerland, May 1997). (12) M.V. Gorenstein, J.B. Li, J. Van Antwerp, and D. Chapman, LCGC 12(10), 768–772 (1994). (13) P.M. Young and M.V. Gorenstein, LCGC 12(11), 832–838 (1994). (14) W.J. Warren, W.A. Stanick, M.V. Gorenstein, and P.M. Young, BioTechniques 18(2), 282–297 (1995). (15) M.E. Swartz, J. Liq. Chrom. 28(7/8), 1253–1263 (2005). (16) M. Swartz and B. Murphy, Am. Lab. 37(3), 22–27 (2005). (17) M. Swartz, Pharm. Formulation Quality 6(5), 40–42 (2004). (18) I.S. Krull and M.E. Swartz, LCGC 15(6) 534–540 (1997). (19) M.E. Swartz and I.S. Krull, Analytical Method Development and Validation (Marcel Dekker, New York, 1997). (20) J. Orr, I.S. Krull, and M.E. Swartz, LCGC 21(7), 626 (2003). (21) J. Orr, M.E. Swartz, and I.S. Krull, LCGC 21(12), 1146 (2003).
|