![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Precision
|
|
Precision is the measure of the degree of repeatability of an analytical method under normal operation and is normally expressed as the percent relative standard deviation for a statistically significant number of samples. According to the ICH, precision should be performed at three different levels: repeatability, intermediate precision, and reproducibility. Repeatability is the results of the method operating over a short time interval under the same conditions (inter-assay precision). It should be determined from a minimum of nine determinations covering the specified range of the procedure (for example, three levels, three repetitions each) or from a minimum of six determinations at 100% of the test or target concentration. Intermediate precision is the results from within lab variations due to random events such as different days, analysts, equipment, etc. In determining intermediate precision, experimental design should be employed so that the effects (if any) of the individual variables can be monitored. |
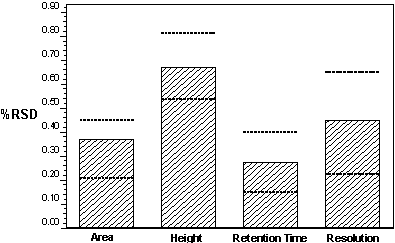
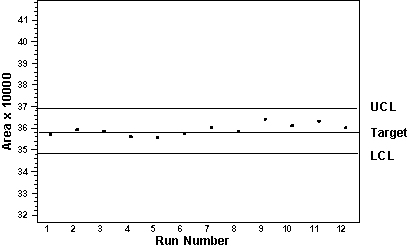
Figure 6: Documenting Precision With Waters® Millennium Chromatography Manager Summary Graphics Bar Plot Custom Report |
Reproducibility refers to the results of
collaborative studies between laboratories.
Documentation in support of precision studies should
include the standard deviation, relative standard
deviation, coefficient of variation, and the
confidence interval. Figure 6 illustrates how custom
graphics in the form of
bar charts can be used to document precision.
|
|
Specificity is the ability to measure accurately and specifically the analyte of interest in the presence of other components that may be expected to be present in the sample matrix. It is a measure of the degree of interference from such things as other active ingredients, excipients, impurities, and degradation products, ensuring that a peak response is due to a single component only. i.e. that no co-elutions exist. Specificity is measured and documented in a separation by the resolution, plate count (efficiency), and tailing factor. Specificity can also be evaluated with modern photodiode array detectors that compare spectra collected across a peak mathematically as an indication of peak homogeneity. ICH also uses the term specificity, and divides it into two separate categories: identification, and assay/impurity tests.
For identification purposes, specificity is
demonstrated by the ability to discriminate between
compounds of closely related structures, or by
comparison to known reference materials. For assay
and impurity tests, specificity is demonstrated by
the resolution of the two closest eluting compounds.
These compounds are usually the major component or
active ingredient and an impurity. If impurities
are available, it must be demonstrated
that the assay is unaffected by the presence of
spikedmaterials (impurities and/or excipients). If
the impurities are not available, the test
results are compared to a second well-characterized
procedure. For assay, the two results are compared.
For impurity tests, the impurity profiles are
compared head-to-head.
|
|
The limit of
detection (LOD) is defined as the lowest
concentration of an analyte in a sample that can
be detected, not quantitated. It is a limit test
that specifies whether or not an analyte is
above or below a certain value. It is expressed
as a concentration at a specified
signal-to-noise ratio, usually two- or
three-to-one. The ICH has recognized the
signal-to-noise ratio convention, but also lists
two other options to determine LOD: visual
non-instrumental methods and a means of
calculating the LOD. Visual non-instrumental
methods may include LOD’s determined by
techniques such as thin layer chromatography
(TLC) or titrations. LOD’s may also be
calculated based on the standard deviation of
the response (SD) and the slope of the
calibration curve (S) at levels approximating
the LOD according to the formula: LOD =
3.3(SD/S). The standard deviation of the
response can be determined based on the standard
deviation of the blank, on the residual standard
deviation of the regression line, or the
standard deviation of y-intercepts of regression
lines. The method used to determine LOD should
be documented and supported, and an appropriate
number of samples should be analyzed at the
limit to validate the level.
|
|
The Limit of
Quantitation (LOQ) is defined as the lowest
concentration of an analyte in a sample that can
be determined with acceptable precision and
accuracy under the stated operational conditions
of the method. Like LOD, LOQ is expressed as a
concentration, with the precision and accuracy
of the measurement also reported. Sometimes a
signal-to-noise ratio of ten-to-one is used to
determine LOQ. This signal-to-noise ratio is a
good rule of thumb, but it should be remembered
that the determination of LOQ is a compromise
between the concentration and the required
precision and accuracy. That is, as the LOQ
concentration level decreases, the precision
increases. If better precision is required, a
higher concentration must be reported for LOQ.
This compromise is dictated by the analytical
method and its intended use. The ICH has
recognized the ten-to-one signal-to-noise ratio
as typical, and also, like LOD, lists the same
two additional options that can be used to
determine LOQ, visual non-instrumental methods
and a means of calculating the LOQ. The
calculation method is again based on the
standard deviation of the response (SD) and the
slope of the calibration curve (S) according to
the formula: LOQ = 10(SD/S). Again, the standard
deviation of the response can be determined
based on the standard deviation of the blank, on
the residual standard deviation of the
regression line, or the standard deviation of
y-intercepts of regression lines.
|
|
Like LOD, the
method used to determine LOQ should be
documented and supported, and an appropriate
number of samples should be analyzed at the
limit to validate the level. One additional
detail should also be considered; both the LOQ
and the LOD can be affected by the
chromatography. Figure 7 shows how efficiency
and peak shape can affect the signal-to-noise
ratio.
|
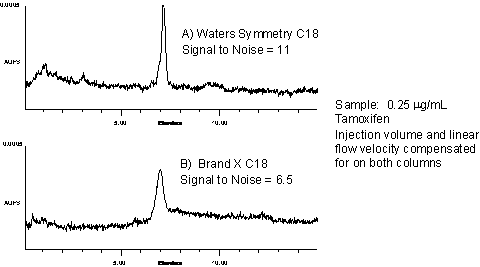
Figure 7: Effect of Peak Shape on LOD/LOQ |
|
Sharper peaks
result in a higher signal-to-noise ratio,
resulting in lower LOQs and LODs. Therefore, the
chromatographic determination of LOQ/LOD should
take into account both the type and age of the
column, which is usually determined over the
course of time as experience with the method
grows.
|
|
Linearity is
the ability of the method to elicit test results
that are directly proportional to analyte
concentration within a given range. Linearity is
generally reported as the variance of the slope
of the regression line. Range is the interval
between the upper and lower levels of analyte
(inclusive) that have been demonstrated to be
determined with precision, accuracy and
linearity using the method as written. The range
is normally expressed in the same units as the
test results obtained by the method. The ICH
guidelines specify a minimum of five
concentration levels, along with certain minimum
specified ranges. For assay, the minimum
specified range is from 80-120% of the target
concentration. For an impurity test, the minimum
range is from the reporting level of each
impurity, to 120% of the specification. (For
toxic or more potent impurities, the range
should be commensurate with the controlled
level.)
For content uniformity testing, the minimum range is
from 70-130% of the test or target concentration,
and for dissolution testing, +/- 20% over the
specified range of the test. That is, in the case of
an extended release product dissolution test, with a
Q-factor of 20% dissolved after six hours, and 80%
dissolved after 24 hours, the range would be 0-100%.
Figure 8 is an example of a linearity (calibration)
plot.
|
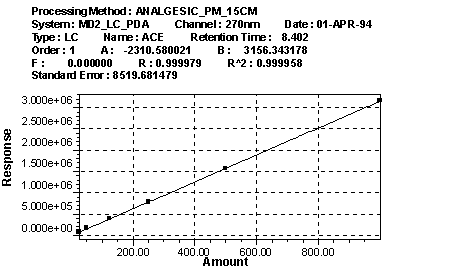
Figure 8: Waters Millennium Chromatography Manager Calibration Plot |
|
Ruggedness,
according to the USP, is the degree of
reproducibility of the results obtained under a
variety of conditions, expressed as %RSD. These
conditions include different laboratories,
analysts, instruments, reagents, days, etc. In
the guideline on definitions and terminology,
the ICH did not address ruggedness specifically.
This apparent omission is really a matter of
semantics, however, as ICH chose instead to
cover the topic of ruggedness as part of
precision, as discussed previously.
|
|
Robustness is
the capacity of a method to remain unaffected by
small deliberate variations in method
parameters. The robustness of a method is
evaluated by varying method parameters such as
percent organic, pH, ionic strength,
temperature, etc., and determining the effect
(if any) on the results of the method. As
documented in the ICH guidelines, robustness
should be considered early in the development of
a method. In addition, if the results of a
method or other measurements are susceptible to
variations in method parameters, these
parameters should be adequately controlled and a
precautionary statement included in the method
documentation.
|